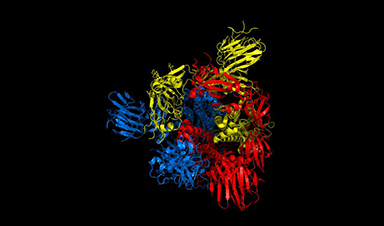
esearchers have detailed a mechanism in the distinctive corona of Covid-19 that could help scientists to rapidly find new treatments for the virus, and quickly test whether existing treatments are likely to work with mutated versions as they develop.
The team, led by the University of Warwick as part of the EUTOPIA community of European universities, have simulated movements in nearly 300 protein structures of the Covid-19 virus spike protein by using computational modelling techniques, in an effort to help identify promising drug targets for the virus.
In a new paper published today (19 February) in the journal Scientific Reports, the team of physicists and life scientists detail the methods they used to model the flexibility and dynamics of all 287 protein structures for the Covid-19 virus, also known as SARS-CoV-2, identified so far. Just like organisms, viruses are composed of proteins, large biomolecules that perform a variety of functions. The scientists believe that one method for treating the virus could be interfering with the mobility of those proteins.
They have made their data, movies and structural information, detailing how the proteins move and how they deform, for all 287 protein structures for Covid-19 that were available at the time of the study, publicly accessible to allow others to investigate potential avenues for treatments.
The researchers focused particular efforts on a part of the virus known as the spike protein, also called the Covid-19 echo domain structure, which forms the extended corona that gives coronaviruses their name. This spike is what allows the virus to attach itself to the ACE2 enzyme in human cell membranes, through which it causes the symptoms of Covid-19.
The spike protein is in fact a homotrimer, or three of the same type of protein combined. By modelling the movements of the proteins in the spike, the researchers identified a ‘hinge’ mechanism that allows the spike to hook onto a cell, and also opens up a tunnel in the virus that is a likely means of delivering the infection to the hooked cell. The scientists suggest that by finding a suitable molecule to block the mechanism – literally, by inserting a suitably sized and shaped molecule – pharmaceutical scientists will be able to quickly identify existing drugs that could be effective against the virus.
Lead author Professor Rudolf Roemer from the Department of Physics at the University of Warwick, who conducted the work while on a sabbatical at CY Cergy-Paris Université, said: “Knowing how this mechanism works is one way in which you can stop the virus, and in our study we are the first to see the detailed movement of opening. Now that you know what the range of this movement is, you can figure out what can block it.
“All those people who are interested in checking whether the protein structures in the virus could be drug targets should be able to examine this and see if the dynamics that we compute are useful to them.
“We couldn’t look closely at all the 287 proteins though in the time available. People should use the motion that we observe as a starting point for their own development of drug targets. If you find an interesting motion for a particular protein structure in our data, you can use that as the basis for further modelling or experimental studies.”
To investigate the proteins’ movements, the scientists used a protein flexibility modelling approach…
Image Credit: University of Warwick
Post by Amanda Scott, NA CEO. Follow her on twitter @tantriclens
Thanks to Heinz V. Hoenen. Follow him on twitter: @HeinzVHoenen
News
According to Researchers, Your Breathing Patterns Could Hold the Key to Better Memory
Breathing synchronizes brain waves that support memory consolidation. A new study from Northwestern Medicine reports that, much like a conductor harmonizes various instruments in an orchestra to create a symphony, breathing synchronizes hippocampal brain waves to [...]
The Hidden Culprit Behind Alzheimer’s Revealed: Microglia Under the Microscope
Researchers at the CUNY Graduate Center have made a groundbreaking discovery in Alzheimer’s disease research, identifying a critical link between cellular stress in the brain and disease progression. Their study focuses on microglia, the brain’s immune [...]

“Mirror Bacteria” Warning: A New Kind of Life Could Pose a Global Threat
Mirror life, a concept involving synthetic organisms with reversed molecular structures, carries significant risks despite its potential for medical advancements. Experts warn that mirror bacteria could escape natural biological controls, potentially evolving to exploit [...]

Lingering Viral Fragments: The Hidden Cause of Long COVID
Long COVID, affecting 5-10% of COVID-19 patients, might be caused by the enduring presence of the virus in the body. Research suggests that viral fragments, possibly live, linger and lead to symptoms. Addressing this involves antiviral treatments, enhanced [...]
Hidden Scars: How COVID Lockdowns Altered Teen Brains Forever
Research from the University of Washington revealed that COVID-19 lockdowns led to accelerated cortical thinning in adolescents, impacting brain development significantly. This effect was more pronounced in females than males, raising concerns about long-term brain health. The study [...]
Simple Blood Test To Detect Dementia Before Symptoms Appear
UCLA researchers have identified placental growth factor (PlGF) as a potential blood biomarker for early detection of cognitive impairment and dementia. High PlGF levels correlate with increased vascular permeability, suggesting its role in the development [...]

Investing Goldman Sachs asks ‘Is curing patients a sustainable business model?’
Goldman Sachs analysts attempted to address a touchy subject for biotech companies, especially those involved in the pioneering “gene therapy” treatment: cures could be bad for business in the long run. “Is curing patients [...]

The risks of reversed chirality: Study highlights dangers of mirror organisms
A groundbreaking study evaluates the feasibility, risks, and ethical considerations of creating mirror bacteria with reversed chirality, highlighting potential threats to health and ecosystems. In a recent study published in Science, a team of researchers [...]

Alarming Mutation in H5N1 Virus Raises Pandemic Red Flags
NIH-funded study concludes that the risk of human infection remains low A recent study published in Science and funded by the National Institutes of Health (NIH) has found that a single alteration in a protein on the surface [...]

Scientists Discover Genetic Changes Linked to Autism, Schizophrenia
The Tbx1 gene influences brain volume and social behavior in autism and schizophrenia, with its deficiency linked to amygdala shrinkage and impaired social incentive evaluation. A study published in Molecular Psychiatry has linked changes in brain [...]

How much permafrost will melt this century, and where will its carbon go?
Among the many things global warming will be melting this century—sea ice, land glaciers and tourist businesses in seaside towns across the world—is permafrost. Lying underneath 15% of the northern hemisphere, permafrost consists of [...]

A Physics Discovery So Strange It’s Changing Quantum Theory
MIT physicists surprised to discover electrons in pentalayer graphene can exhibit fractional charge. New theoretical research from MIT physicists explains how it could work, suggesting that electron interactions in confined two-dimensional spaces lead to novel quantum states, [...]

Inside the Nano-Universe: New 3D X-Ray Imaging Transforms Material Science
A cutting-edge X-ray method reveals the 3D orientation of nanoscale material structures, offering fresh insights into their functionality. Researchers at the Swiss Light Source (SLS) have developed a groundbreaking technique called X-ray linear dichroic orientation tomography [...]
X-chromosome study reveals hidden genetic links to Alzheimer’s disease
Despite decades of research, the X-chromosome’s impact on Alzheimer’s was largely ignored until now. Explore how seven newly discovered genetic loci could revolutionize our understanding of the disease. Conventional investigations of the genetic contributors [...]

The Unresolved Puzzle of Long COVID: 30% of Young People Still Suffer After Two Years
A UCL study found that 70% of young people with long Covid recovered within 24 months, but recovery was less likely among older teenagers, females, and those from deprived backgrounds. Researchers emphasized the need [...]

Needle-Free: New Nano-Vaccine Effective Against All COVID-19 Variants
A new nano-vaccine developed by TAU and the University of Lisbon offers a needle-free, room-temperature-storable solution against COVID-19, targeting all key variants effectively. Professor Ronit Satchi-Fainaro’s lab at Tel Aviv University’s Faculty of Medical and [...]




